更新时间:2016-04-22 17:03 浏览: 次 作者:admin 文章来源:未知
随着社会的进步,人们的生活条件变得越来越好,心血管疾病也开始肆意盛行起来。据相关报道指出,心血管疾病中,冠状动脉疾病(CAD)最为流行,而且是发达国家死亡的最大原因。根据DATASUS统计,巴西每年大约有100万的医院葬礼起因于心血管疾病。因此,如何治疗动脉粥样硬化早已成为医学研究热点的重中之重。
动脉粥样硬化(atherosclerosis)是动脉硬化的血管病中最常见也是最重要的一种。研究表明,各种动脉硬化症都显示出一定的共性,即动脉管壁增厚、变硬、失去弹性和管腔缩小。导致动脉粥样硬化的因素很多,而且错综复杂,其中重要的两个因子就是胆固醇和脂蛋白。
导致动脉粥样硬化的机理:
1、LDL透过内皮细胞深入内皮细胞间隙,单核细胞迁入内膜,此即最早期。
2、Ox-LDL与巨噬细胞的清道夫受体结合而被摄取,形成巨噬源性泡沫细胞,对应病理变化中的脂纹。
3、动脉中膜的血管平滑肌细胞(SMC)迁入内膜,吞噬脂质形成肌源性泡沫细胞,增生迁移形成纤维帽,对应病理变化中的纤维斑块。
4、Ox-LDL使上述两种泡沫细胞坏死崩解,形成糜粥样坏死物,粥样斑块形成。对应病理变化中的粥样斑块。
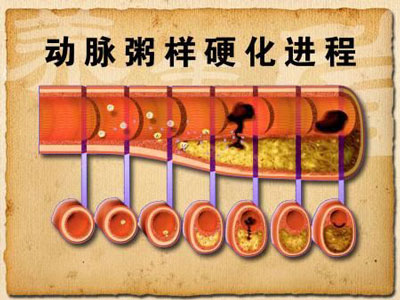
动脉硬化是进行式,不是到此为止。继发性病变有斑块内出血、斑块破裂及局部血栓形成(称为粥样硬化-血栓形成,atherosclerosis-thrombosis)。粥样硬化斑块中脂质及结缔组织的含量决定斑块的稳定性以及是否易导致急性缺血事件发生。
斑块中包含大比例的巨噬细胞相对平滑肌细胞SMCs更不稳定,尤其是那些ACTA2+纤维帽薄弱被认为是组成主要的表型调整型的SMCs。对于动脉粥样硬化斑块中细胞系的起源有广泛的歧义。这些歧义主要基于胆固醇处理后,体外培养的SMCs下调了SMC markers并活化了巨噬细胞markers。已知的损伤中含有的因子如凝血酶处理后,巨噬细胞会激活SMC的基因。而最令人信服的证据是对交叉性骨髓移植研究,在人类进展性冠脉损伤中,SMCs和巨噬细胞通常不能鉴别。这些研究都表明ACTA2+细胞是造血干细胞来源而不是SMC来源。且与之一致的是,Apoe完全缺失小鼠,表达SMC markers的细胞实质部分是HSC来源而不是SMC来源。相反的广泛的证据表明Apoe完全缺失小鼠许多SMC衍生细胞缺乏可检测的传统的SMC marker和/或激活巨噬细胞marker的表达。

尽管几十年的动脉粥样硬化的研究,我们仍不知道斑块内的哪种细胞是SMC衍生的,并且在何种程度上促进斑块的发病机理。主要的歧义在于鉴定动脉粥样硬化斑块中某种细胞是SMC衍生还是巨噬细胞衍生,而解释该歧义关键需要解决以下几个问题。
1、 斑块内的SMCs,巨噬细胞,和其他细胞类型的表型调整是如何调节的?
2、 这些表型调整细胞的功能是什么?
3、 这些表型调整如何影响整体的疾病发病机理。
为了解决这些问题,我们培育除出了动脉粥样硬化易发的Apoe缺失小鼠,用这种方法,我们可以追踪SMCs和研究SMC特异敲除干细胞多潜能基因(KLF4)的效果。Krüppel样因子4(Krüppel-like factor4,KLF4)是含有3个锌指结构的转录因子,在细胞微环境中参与不同的细胞信号网络,调控靶基因的转录活化或抑制,与细胞的增殖分化、诱导型多能干细胞的生成有关,具有癌基因和抑癌基因、促炎和抗炎等双向调节功能。
我们选择研究KLF4的作用,是因为我们和其他人前期证明了这一转录因子在发育过程中,颈动脉结扎损伤,体外培养SMCs经血小板衍生生长因子(PDGF)-BB,PDGF-DD或者氧化磷脂处理后,调节SMCs的表型调整方面起到了重要作用。
实验结果显示约82%的SMCs(YFP+DAPI+ cells)为ACTA2阴性,说明斑块中的多数SMCs不能通过传统的SMCmarkers来识别,表型调整的SMCs(YFP+ACTA2−DAPI+)组成了约30%的斑块内的总细胞。这一结果远超过了以前基于ACTA2免疫染色来估计的调整型SMCs的含量。
动脉粥样硬化斑块中的SMCs表达巨噬细胞,MSCs和肌成纤维细胞的marker。根据我们的结果,推测出斑块内SMC衍生细胞的分配比为30%巨噬样细胞(YFP+ACTA2−LGALS3+),7%的MSC样细胞(YFP+ACTA2−SCA1+),12%肌成纤维样细胞(YFP+ACTA2+PDGFR+),和32-51%不确定细胞类型(YFP+ACTA2−LGALS3−SCA1−)。另外,进展性动脉粥样硬化斑块中36% LGALS3+细胞是YFP+,说明根据以前的研究被分类到巨噬细胞的约1/3的细胞,实际上起源于SMCs而不是髓系细胞。但是尽管动脉粥样硬化的SMC衍生细胞表达了MSCs的多种markers,这些细胞并不表现出多能性,也不表现出MSCs的功能属性。
动脉粥样硬化中的SMCs表达巨噬细胞marker CD68.原位杂交接近结扎实验(ISH-PLA)由本实验室建立,这个技术通过检测Myh11启动子的组蛋白H3K4diMe (PLA+)确定组织中表型调整的SMCs。当RAW264.7鼠巨噬细胞或人单核细胞暴露在POVPC下时(LDL的氧化产物),会激活单核细胞/巨噬细胞,抑制Myh11启动子的组蛋白H3K4diMe。人冠状动脉硬化斑块染色CD68和ACTA2,ISH-PLA检测Myh11启动子的组蛋白H3K4diMe,18%的CD68+细胞PLA+,表明它们是SMC起源的。
KLF4在调节SMC表型和斑块发病机理中起关键作用。以前没有证据证明SMC表型调整是KLF4依赖的。但是我们观察到动脉粥样硬化模型鼠的头臂动脉中大量YFP+细胞表达KLF4。我们培育了SMC特异性KLF4敲除的SMCYFP+/+Apoe−/− 鼠:SMCYFP+/+Klf4∆/∆Apoe−/− 鼠。发现与对照组(SMCYFP+/+Klf4+/+Apoe−/−)相比,约50%斑块范围缩小,增加了斑块的稳定性及纤维帽中ACTA2+细胞的数量,下调了LGALS3+细胞的数量。对SMC表型调整方面,53%巨噬样SMCs(YFP+LGALS3+/YFP+)的下降,70%MSC样SMCs(YFP+SCA1+/YFP+)的下降。 流式分析也得到一致结果。SMCYFP+/+Klf4∆/∆Apoe−/− 鼠表现出纤维帽中ACTA2+细胞增加,而SMC衍生细胞的增殖下降,YFP+SMC凋亡与对照组相比显著下降。
KLF4 调节SMCs的表型调整和功能属性。ChIP—seq分析发现西方饮食Apoe缺失小鼠,KLF4结合Acta2, Tagln, Myh11, andCnn1内生启动子显著增加。ISH-PLA分析,在Apoe缺陷小鼠的斑块个体表型调整的SMCs中,KLF4结合Tagln启动子。SMC marker基因表达的共同抑制是KLF4直接结合到SMC marker基因的启动子上实现的。KLF4可以作为转录抑制剂或激活剂,依赖于细胞的类型和基因的位置。为了更好的定义介导SMC表型的KLF4靶基因的全部内容,我们运用ChIP—seq分析SMCYFP+/+Klf4+/+Apoe−/− versus SMCYFP+/+Klf4∆/∆Apoe−/−小鼠。鉴定了869个KLF4的靶基因。不仅得出KLF4与SMC smarker基因的结合,还发现了KLF4与调节途径基因的结合,包括了吞噬作用,凋亡,细胞迁移,和炎症基因家族。这些推测的KLF4的靶基因可能有益于SMC特异的KLF4缺失缩小损害范围和斑块发病机理。
同时,文章还检测了整体KLF4基因的敲除改变斑块发病机理。发现纯合敲除的小鼠,不能长期存活,主要跟小鼠的体重严重下降及皮肤损害有关。而杂合敲除的小鼠则表现为斑块内出血下降,凋亡减少和细胞增殖。因此一条KLF4等位基因的缺失,由于形成更小更稳定的斑块对斑块的发展是有益的。
我们发现在选择性KLF4的缺失时,可以导致损伤范围减小,增加纤维帽的厚度,SMC衍生的巨噬样细胞,MSC样细胞含量的大幅度下降,但是增加了纤维帽中ACTA2+细胞的分数。另外,我们发现培养的SMCs胆固醇加载会诱导KLF4依赖的巨噬marker和MSC marker的激活,促炎因子的表达,增加吞噬行为。最后,我们体内ChIP—seq分析鉴定大于800个推测的KLF4在SMCs中的调节基因,包括许多相关的促炎过程。综合来讲,这些结果提供了令人信服的证据:SMCs表型调整在损害发展,斑块组成,稳定性方面中有关键的作用。因此,瞄准促进SMC表型有益的变化的治疗方式可能成为处理进展性动脉粥样硬化的可行的方法。然而,KLF4在SMCs中的缺失是如何导致全面的损害范围减小的,以及斑块稳定性方面多种改变是如何发生的仍然是一个关键的问题。最近,Fisher实验室发现胆固醇加载后,SMCs表达巨噬细胞marker,但是芯片数据显示这些细胞的主要成分与典型的单核细胞,巨噬细胞和树突细胞明显的不同。这些细胞的吞噬能力降低。而我们也发现SMC衍生的MSC样细胞功能异常。
有研究表明,实验动物的动脉粥样硬化病变,在用药物治疗和停止致动脉粥样硬化饲料一段时间后,病变甚至可完全消退。在人体经血管造影或腔内超声检查证实,控制和治疗各危险因素一段时间后,较早期的动脉粥样硬化病变可部分消退。未来研究的关键挑战是鉴定进展动脉粥样硬化损害中调节SMCs表型的的环境诱因,以及损害中其他主要的细胞类型,并这些如何在治疗上降低斑块负担和增加斑块稳定性。
(来源:百替生物,作者:Oprah)