更新时间:2016-04-22 17:00 浏览: 次 作者:admin 文章来源:未知
近期Nature Medicine一篇研究中,研究人员系统性地分析了来自8000多例癌症患者已经测序过的基因组编码序列的复杂插入和缺失突变图谱,从中发现了285个癌症相关的基因存在复杂插入和缺失突变。在先前报道的2199个样本中,几乎所有的这些新发现的复杂插入和缺失突变都被忽略(81.1%)或错误注释(17.6%)。
高通量测序技术将医学研究推向了一个史无前例的高度。通过此项技术,人们累积了大量、复杂的数据,如单个碱基突变,小片段插入和缺失或结构型突变等等。
从Sanger法测序到下一代测序技术,人们逐渐能够在极短的时间,完成诸如人类全基因组测序这类浩瀚的工程。然而,通过对癌症患者测序结果的进一步分析。这些“价值连城”的大数据,其利用率,可能不到20%。造成这一现象的原因主要是没有有效的工具,从数以百万的碱基中,提高数据的分辨率。
研究人员构建了Pindel-C的新算法,系统性地分析了来自8000多例癌症患者已经测序过的基因组编码序列的复杂插入和缺失突变图谱。
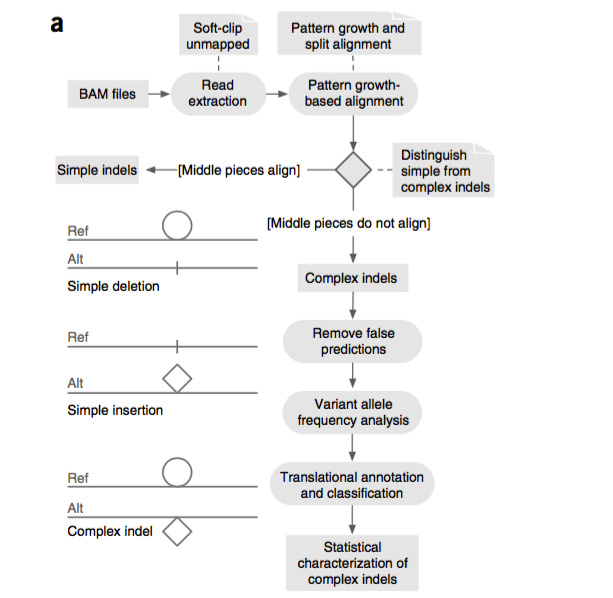
研究人员发现了285个癌症相关的基因(如PIK3R1、TP53、ARID1A、GATA3以及KMT2D等)存在复杂插入和缺失突变。在先前报道的2199个样本中,几乎所有的这些新发现的复杂插入和缺失突变都被忽略(81.1%)或错误注释(17.6%)。
进一步发现,PIK3R1和EGFR常发生插入和缺失突变,而大部分VHL、GATA3、TP53、ARID1A、PTEN和ATRX基因存在移码突变。此外,复杂插入和缺失突变还表现出较强的组织特异性(例如,肾癌中常发生VHL突变,乳腺癌病例中经常出现GATA3突变)。
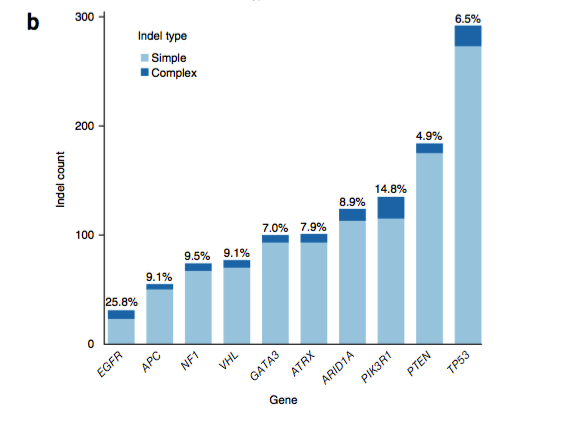
这些复杂的突变经常聚集在重要的癌基因中,被归结为随机改变。由于一些插入缺失突变存在于已有药物设计来对抗突变效应的基因中,检测到他们具有重要的临床意义。
研究人员发现,在与肺癌相关的EGFR基因中发现了一些复杂插入缺失突变,如果这样的插入缺失存在于这一基因中,患者有可能会从EFGR抑制剂如埃罗替尼(erlotinib)中受益,不管肿瘤类型为何。
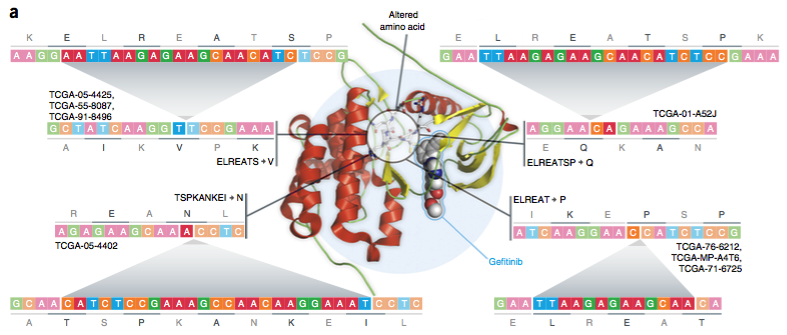
研究人员还在KIT基因中发现了一些复杂插入缺失,KIT似乎在黑色素瘤中发挥作用。分析结果表明,具有KIT复杂插入缺失的患者将从伊马替尼、苏尼替尼和索拉菲尼中受益,这些药物靶向了这一基因的突变。
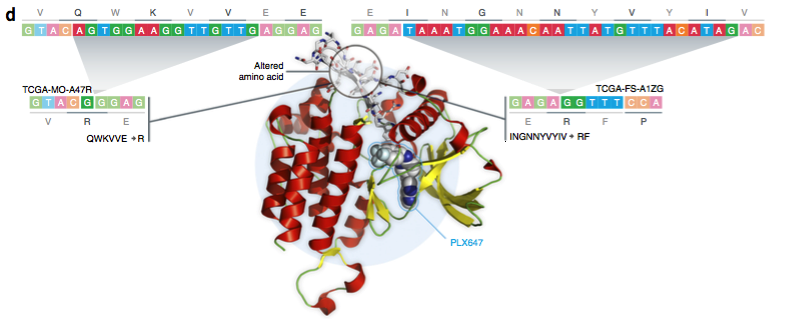
当前的基因组分析方法会系统性地错过检测癌症患者肿瘤中某种类型的复杂插入缺失突变。借助Pindel-C类似高质量的算法,我们能够将它们一一找寻出来并进行标注与分析。更为详细的研究结果,将能指导癌症个性化治疗。